 2023, Vol. 50
2023, Vol. 50



新生儿慢性肉芽肿病(chronic granulomatous disease,CGD)是一种少见的原发免疫缺陷病,发生率约占活产婴儿的1~3/20万[1-2]。新生儿起病更少见,且临床症状多不典型,而早期识别诊断有助于该病治疗,对改善预后有重要意义。现报道复旦大学附属儿科医院1例新生儿期起病的慢性肉芽肿病,以提高临床上对该病的认识,并提高诊断效率。
病例资料 患儿,男,因反复气促、发热1个月入院,第2胎第2产,胎龄40+2周,顺产出生,出生体重3 050 g,出生无窒息史,Apgar评分1~5 min为10~10分。出生后20天开始发热,最高38 ℃,伴咳嗽,2天后至当地医院就诊,查血常规示:白细胞34.8×109/L,中性粒细胞百分比63%,血小板308×109/L。胸片提示新生儿肺炎,先后给予红霉素、头孢哌酮钠舒巴坦钠、美罗培南、利奈唑胺等多种抗生素抗感染治疗,丙种球蛋白输注支持治疗。患儿呼吸困难,予鼻导管吸氧治疗。胸部CT示:两肺弥漫结节、团块影伴纵膈淋巴结肿大,骨髓穿刺涂片细胞学检查无异常,支气管镜检查提示支气管内膜炎。加用卡泊芬净抗真菌治疗,病情无好转,仍有发热,伴气促、咳嗽。出生后50天,转诊至复旦大学附属儿科医院。患儿出生后接种乙肝疫苗,未接种卡介苗。新生儿疾病筛查正常。入院体重3 760 g,体温38 ℃,血压85/51 mmHg(1 mmHg=0.133 kPa,下同),脉搏155次/min,呼吸频率50次/min。鼻导管吸氧下,经皮血氧饱和度稳定,神志清晰,前囟平软,双肺呼吸音粗,可闻及痰鸣音,心音有力,心律齐,未闻及杂音,腹部平软,肝脾无肿大,肠鸣音正常,四肢肌力肌张力正常。
入院检验结果 血常规示:白细胞(21.4~41.8)×109/L,血红蛋白91~128 g/L,血小板为(533~860)×109/L,C反应蛋白(C-reactive protein,CRP)27.9~133.2 mg/L。尿液分析、粪常规、血气分析、凝血全套、生化常规、血培养、痰培养、呼吸道病原体检测、降钙素原(procalcitonin,PCT)、肠道病毒、自身抗体13项(包括抗双链DNA抗体、抗核抗体、抗Sm抗体等)、真菌葡聚糖(G试验)、半乳甘露聚糖(GM试验)、TORCH、真菌培养、EBV-DNA及CMV-DNA定量测定、脑脊液等检测均未见明显异常。血清铁蛋白829.7 ng/mL,免疫球蛋白IgG 12.0 g/L(正常2.75~7.50 g/L),IgM 0.78 g/L(正常0.1~0.7 g/L),余未见异常。流式细胞分析示:CD4+T细胞比例50.40%(正常29%~36%),CD8+T细胞比例14.56%(正常24%~34%),CD19+B细胞比例25.70%(正常14%~21%),CD16+和CD56+NK细胞比例5.59%(正常11%~23%),gp91蛋白不表达。
胸部影像学检查结果 如图 1所示,胸部X线片示:两肺多发结节及团块影,部分融合;胸部增强CT示:两肺多发结节、肿块,增强后不均匀强化,纵膈、右肺门淋巴结肿大。

|
| A: Chest radiograph showed multiple nodules and mass shadows; B-D: Enhanced CT showed multi focal masses or nodules, mediastinal and right hilar lymphadenopathy. 图 1 CGD患儿胸部X线片和增强CT检查结果 Fig 1 Chest X-ray and contrast-enhanced CT findings of the newborn with CGD |
中性粒细胞呼吸爆发功能检测结果 如图 2所示,患儿中性粒细胞刺激指数2.4(正常 > 100)显著异常,母亲中性粒细胞刺激指数124(正常)。

|
| The stimulation index of neutrophils in the neonate and his mother were 2.4 and 124, respectively. The stimulation index of neutrophils in the neonate was remarkable abnormality. 图 2 患儿及母亲中性粒细胞呼吸爆发功能的流式细胞检测结果 Fig 2 Flow cytometric assay for respiratory burst function of neutrophils in the neonate and his mother |
基因检测结果 父母及患儿家系基因检测结果显示:患儿CYBB基因在染色体位置chrX:37655385;c.665(exon6)A > G(p.H222P)错义突变,来源于母亲(图 3),明确诊断新生儿CGD。

|
| 图 3 患儿及父母CYBB基因测序图 Fig 3 Sequencing of gene CYBB of the neonate and his parents |
治疗 患儿入院后,给予万古霉素、头孢吡肟、氟康唑抗感染治疗,痰、尿、血、脑脊液培养均为阴性。怀疑CGD后,根据肺部影像学表现,临床考虑霉菌性肺炎,改为伏立康唑抗真菌治疗,并转诊至临床免疫科进一步治疗。在抗真菌治疗后,患儿肺部感染及结节病灶好转,后多次因感染入院治疗,现等待造血干细胞移植。
讨论 新生儿CGD是一种罕见的原发性吞噬细胞免疫缺陷病,由还原型烟酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotidephosphate,NADPH)氧化酶复合物缺陷所致。NADPH氧化酶复合物有5个亚单位结构,包括gp91phox、p22phox、p47phox、p67phox、p40phox,分别由CYBB、CYBA、NCF1、NCF2、NCF4等5个基因所编码[3-5]。CYBB基因缺陷是CGD最常见的类型,发病机制为NADPH氧化酶缺陷导致中性粒细胞、单核细胞、巨噬细胞、嗜酸性粒细胞产生超氧阴离子及其他活性氧中间体的能力受损[6]。CGD发病率低,临床主要表现为反复细菌、真菌感染,过度炎症反应等[7]。
新生儿CGD的早期诊断对于积极寻求治疗方案至关重要。CGD的早期诊断主要依赖临床症状、常规检查检验、针对性功能实验、蛋白质水平检测和基因诊断等。本例临床表现为对抗生素等常规治疗反应欠佳的严重感染。胸片和胸部CT影像学、功能实验、基因检测可为CGD诊断提供重要依据。
CGD肺部真菌感染的肺部影像学检查有特异性。本例患儿主要表现为结节样改变,结节融合成片。文献报道的新生儿病例CT影像检查也以类似征象为主[8-10]。赵晶等[11]报道30例儿童CGD的胸部CT影像学,主要表现为实变、结节、团块、空洞、间质改变等,结节与团块样改变高度提示曲霉菌感染。在病原菌检查不明确时,CT对提示曲霉菌感染具有重要参考价值。本例患儿多次痰培养均为阴性,给予大扶康治疗患儿肺部病变改善不明显,经验性改为伏立康唑治疗后,肺部病变逐渐好转,提示曲霉菌感染可能性大。值得注意的是,该患儿真菌培养阴性,而且G试验和GM试验均为阴性,提示常规真菌检测手段在CGD患儿真菌感染中假阴性可能较高。
中性粒细胞爆发功能检测是CGD针对性功能实验。正常情况下,中性粒细胞通过一系列氧气依赖和非氧气依赖途径抵御病原入侵。中性粒细胞吞噬病原后,消耗大量氧气,这一过程由NADPH氧化酶复合物介导,产生活性氧(reactive oxygen species,ROS)。NADPH氧化酶复合物氧气消耗并产生超氧阴离子及其他活性氧中间物的过程称为呼吸爆发[12]。流式细胞仪-DHR分析是检测活性氧的主要方法,可用于CGD诊断[13]。刺激指数用于评价ROS产生能力,正常人群平均值为127.9(85.2~264.4)[14]。本例患儿刺激指数仅为2.4。周钦华等[15]报道138例儿童CGD病例刺指数为2.69(0.8~60.5)。gp91蛋白检测结果显示,患儿不表达gp91,从功能学上提示CYBB基因缺陷。
近年来,基因检测已成为CGD诊断的重要手段。CYBB基因缺陷是CGD基因缺陷的重要类型,占比达65.0%~89.1%[15]。该基因位于X染色体上,因此母亲多为携带者,但不发病。本例患儿有CYBB基因缺陷,为错义突变,来源于母亲。文献报道CYBB基因突变80%以上来源于母亲,另10%为自身新发突变[16]。基因突变类型中,超过一半为单基因替换、小缺失或基因插入约占1/4[17]。
CGD早期诊断后,因感染入院的患儿治疗方案以控制感染为主,控制急性期感染后需要长期服用药物以预防感染,如复方磺胺甲噁唑、伊曲康唑等预防细菌、真菌感染。新生儿病例及对复方磺胺甲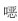
本文报道了1例CGD患儿,梳理了CGD现有诊断方案,从临床、功能及基因分析全面探讨相关诊断方案的临床意义,以期为CGD的早期诊断提供参考。CT影像学显示结节、团块等特征性表现提示肺部曲霉菌感染可能;中性粒细胞呼吸爆发试验、gp91蛋白检测、基因检测等是诊断CGD的重要手段。
作者贡献声明 高瑞伟 资料收集,病例管理,论文撰写。张鹏 资料收集,论文撰写。张蓉 病情分析,论文修订。曹云疾 病诊疗,论文修订。周建国 疾病诊疗,研究指导,论文修订。
利益冲突声明 所有作者均声明不存在利益冲突。
| [1] |
MILADINOVIC M, WITTEKINDT B, FISCHER S, et al. Case report: symptomatic chronic granulomatous disease in the newborn[J]. Front Immunol, 2021, 12: 663883.
[DOI]
|
| [2] |
WOLACH B, GAVRIELI R, DE BOER M, et al. Chronic granulomatous disease: clinical, functional, molecular, and genetic studies. The Israeli experience with 84 patients[J]. Am J Hematol, 2017, 92(1): 28-36.
[DOI]
|
| [3] |
DINAUER MC, ORKIN SH, BROWN R, et al. The glycoprotein encoded by the X-linked chronic granulomatous disease locus is a component of the neutrophil cytochrome B complex[J]. Nature, 1987, 327(6124): 717-720.
[DOI]
|
| [4] |
DINAUER MC, PIERCE EA, BRUNS GA, et al. Human neutrophil cytochrome B light chain (p22-phox). Gene structure, chromosomal location, and mutations in cytochrome-negative autosomal recessive chronic granulomatous disease[J]. J Clin Invest, 1990, 86(5): 1729-1737.
[DOI]
|
| [5] |
ROOS D, VAN LK, HSU AP, et al. Hematologically important mutations: the autosomal forms of chronic granulomatous disease (third update)[J]. Blood Cells Mol Dis, 2021, 92: 102596.
[DOI]
|
| [6] |
BOUSFIHA A, JEDDANE L, PICARD C, et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification[J]. J Clin Immunol, 2020, 40(1): 66-81.
[DOI]
|
| [7] |
YU HH, YANG YH, CHIANG BL. Chronic granulomatous disease: a comprehensive review[J]. Clin Rev Allergy Immunol, 2021, 61(2): 101-113.
[DOI]
|
| [8] |
周炎娟, 康文清, 张耀东, 等. 新生儿慢性肉芽肿4例临床诊治体会[J]. 广东医学, 2020, 41(10): 1041-1046. [CNKI]
|
| [9] |
罗丁珍, 卢文青. 新生儿慢性肉芽肿病一例[J]. 中华新生儿科杂志(中英文), 2019, 34(6): 466-467. [DOI]
|
| [10] |
王玲, 陈晓文, 欧阳珊, 等. 新生儿慢性肉芽肿病合并曲霉菌感染一例报告并文献复习[J]. 中华新生儿科杂志(中英文), 2019, 34(1): 42-46. |
| [11] |
赵晶, 彭芸, 贺建新. 儿童慢性肉芽肿病胸部CT表现[J]. 医学影像学杂志, 2012, 22(3): 359-363. [CNKI]
|
| [12] |
WITKO-SARSAT V, RIEU P, DESCAMPS-LATSCHA B, et al. Neutrophils: molecules, functions and pathophysiological aspects[J]. Lab Invest, 2000, 80(5): 617-653.
[DOI]
|
| [13] |
EMMENDORFFER A, NAKAMURA M, ROTHE G, et al. Evaluation of flow cytometric methods for diagnosis of chronic granulomatous disease variants under routine laboratory conditions[J]. Cytometry, 1994, 18(3): 147-155.
[DOI]
|
| [14] |
VOWELLS SJ, SEKHSARIA S, MALECH HL, et al. Flow cytometric analysis of the granulocyte respiratory burst: a comparison study of fluorescent probes[J]. J Immunol Methods, 1995, 178(1): 89-97.
[DOI]
|
| [15] |
周钦华, 刘丹如, 王莹, 等. 慢性肉芽肿病的实验室诊断[J]. 中华儿科杂志, 2016, 54(5): 337-343. [DOI]
|
| [16] |
SEGAL BH, LETO TL, GALLIN JI, et al. Genetic, biochemical, and clinical features of chronic granulomatous disease[J]. Medicine (Baltimore), 2000, 79(3): 170-200.
|
| [17] |
STASIA MJ, LI XJ. Genetics and immunopathology of chronic granulomatous disease[J]. Semin Immunopathol, 2008, 30(3): 209-235.
|
| [18] |
禹定乐, 王文建, 郑跃杰, 等. 儿童慢性肉芽肿病研究进展[J]. 中华实用儿科临床杂志, 2020, 35(11): 877-880. [CNKI]
|
| [19] |
DEDIEU C, ALBERT MH, MAHLAOUI N, et al. Outcome of chronic granulomatous disease-conventional treatment vs stem cell transplantation[J]. Pediatr Allergy Immunol, 2021, 32(3): 576-585.
|
| [20] |
CHIESA R, WANG J, BLOK HJ, et al. Hematopoietic cell transplantation in chronic granulomatous disease: a study of 712 children and adults[J]. Blood, 2020, 136(10): 1201-1211.
|
| [21] |
GUNGOR T, CHIESA R. Cellular therapies in chronic granulomatous disease[J]. Front Pediatr, 2020, 8: 327.
|
| [22] |
KOHN DB, BOOTH C, KANG EM, et al. Lentiviral gene therapy for X-linked chronic granulomatous disease[J]. Nat Med, 2020, 26(2): 200-206.
|
| [23] |
唐湘凤, 卢伟, 井远方, 等. 单倍体造血干细胞联合第三方脐血移植治疗慢性肉芽肿临床研究[J]. 中国当代儿科杂志, 2019, 21(6): 552-557. [CNKI]
|
| [24] |
李智慧, 王旖旎, 王昭. 单倍体异基因造血干细胞移植治疗慢性肉芽肿病合并慢性活动性EB病毒感染一例并文献复习[J]. 白血病·淋巴瘤, 2020, 29(10): 605-609. |
| [25] |
钱晓文, 翟晓文, 江文晋, 等. 非血缘脐血干细胞移植治疗慢性肉芽肿病[C]. 珠海: 中华医学会第二十一次全国儿科学术大会论文集, 2016: 120.
|